
Введение. С 2005 по 2013 г. в Республике Дагестан нами проводились клинико-эпидемиологическое и молекулярно – генетическое исследования наследственных нервно-мышечных заболеваний (ННМЗ) с созданием регистра ННМЗ. Среди них конечностно - поясные прогрессирующие мышечные дистрофии составили 12,8% [1].
В литературе по данным некоторых авторов распространенность конечностно - поясной прогрессирующей мышечной дистрофии (КППМД) колеблется 1,76-5,0 на 100 тыс. человек населения [2]. С.Н. Иллариошкин в своих работах показал генетическую гетерогенность и клинический полиморфизм данной формы заболевания [3].
В ходе проведения данного исследования нами выявлен горный изолят, расположенный на высоте примерно 1600 м над уровнем моря с численностью населения 2909 человек, в котором выявлено накопление прогрессирующей мышечной дистрофии конечностно-поясной формы (КППМД).
Цель. Верификация клинического и молекулярно-генетического диагноза ПМД и определение частоты гетерозиготного носительства в горном изоляте мутации гена дисферлин (DYSF)*.
Материалы и методы. В работе использован созданный в Дагестане регистр наследственных нервно-мышечных заболеваний «Нейрорегистр Дагестана» на основе платформы для разработки диагностических программ xGen IDS с применением языка программирования Visual Basic в среде разработки MS Visual Studio 98.
Молекулярно-генетическое исследование пациентов и фенотипически здоровых жителей горного изолята проводили в лаборатории ДНК-диагностики ФГБНУ «МГНЦ».
Результаты. В исследованном горном изоляте выявлено 18 пациентов с ККПМД. Распространенность данной патологии в изоляте составила 5,1 на 1 тыс. человек [4].
Клиническими критериями для верификации нозологической формы КППМД являлись: прогрессирующее течение заболевания, нарушения походки (переваливающаяся), приемы Говерса (подъем «лесенкой» или по самому себе из положения сидя на полу), мышечная слабость и гипертрофия мышц тазового и плечевого поясов, гиперлордоз в поясничном отделе, наличие контрактур преимущественно в голеностопных суставах, деформация грудной клетки, затруднение ходьбы на пятках, псевдогипертрофии мышц, первично-мышечный характер поражения при ЭНМГ исследовании, повышение уровня КФК в крови, что соответствовало литературным данным.
Таким образом, после анализа основных симптомов заболевания в рассматриваемой выборке пациентов нами выделены две клинические группы КППМД с АР типом наследования, представленные в табл. 1.
Таблица 1. КППМД
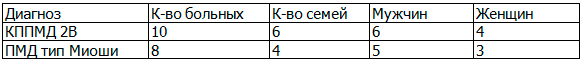
Из представленной таблицы 1 следует, что в обеих группах среди пациентов преобладают лица мужского пола.
КППМД 2В. КППМД 2В диагностирована у 10 пациентов в 6 семьях. В исследованных семьях кровнородственный брак не зарегистрирован. Однако, учитывая длительную изолированность населения выявленного горного изолята можно предположить наличие дальнего родства (6-8 степени) между семьями, и как следствие возможность схожих генотипов. Возраст дебюта заболевания составил 11-21 год. У 100% пациентов болезнь начиналась со слабости в проксимальных отделах одновременно ног и рук с преимущественным поражением передних групп мышц, арефлексией сухожильных рефлексов. Также во всех случаях выявлено умеренное повышение уровня креатинфосфокиназы (КФК) в пределах 1200-2700 Ед/л. При проведении электронейромиографического исследования выявлялся первично-мышечный характер поражения. У 6 пациентов (60%) отмечались псевдогипертрофии икроножных мышц.
Таким образом, в ходе проведения дифференциальной диагностики данной формы заболевания дифференциально-диагностическими критериями являлись: нормальное раннее развитие, начало болезни во втором десятилетии жизни, затруднение ходьбы на пятках, преобладание слабости в задних группах мышц ног, иногда псевдогипертрофии икроножных мышц, а также умеренный уровень КФК.
ПМДТМ. Дистальный тип Миоши (ПМДТМ) диагностирован у 8 пациентов в четырех семьях. Во всех семьях зарегистрированы кровнородственные браки 2-3-4 степени родства. Возраст манифестации с 15-21 лет. У пациентов наблюдались атрофии задней группы мышц дистальных отделов ног и мышц предплечий, ретракция ахилловых сухожилий. Болезнь дебютировала с миалгии и сопровождалась значительным повышением уровня КФК (от 7000 до 31200 Ед/л). У отдельных пациентов в доклинической стадии отмечалось повышение в крови уровня АЛТ (аланинтрансаминаза) и АСТ (аспартатаминотрансфераза) в пределах 78-124 Ед/л.
На МРТ у пациентов с ПМДДМ выявлены атрофические изменения преимущественно в медиальных головках икроножных мышц с замещением жировой клетчаткой; в меньшей степени такие изменения наблюдались в передних большеберцовых и задних бедренных мышцах. В то время как у пациентов с ПМД 2В типа в большей степени страдали мышцы бедер [5].
Диагностическими критериями являлись: слабость в дистальных отделах ног, присоединение по мере прогрессирования болезни поражения мышц надплечий, а также крайне высокий уровень КФК, что было в 50-200 раз больше нормальных значений [6].
Таким образом, в исследуемых семьях были определены дифференциальные клинико-диагностические критерии КППМД 2В и ПМДТМ, представленные в табл. 2.
Таблица 2. Клинические критерии КППМД 2В и ПМДТМ
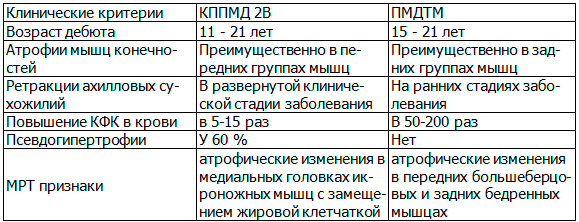
Как видно из табл. 2 в клинической картине КППМД 2В и ПМДДМ имеются ряд характерных клинических и лабораторно-диагностических особенностей, позволяющих дифференцировать данные патологии. Так для КППМД 2В характерен более ранний возраст дебюта заболевания, преимущественное поражение передних групп мышц конечностей, у большей части пациентов имеются псевдогипертрофии икроножных мышц, при этом КФК в крови повышена умеренно. Для дистального типа Миоши значимы более поздний возраст дебюта 2-3 десятилетие, поражение задней группы мышц конечностей, деформации голеностопных суставов на раннем этапе заболевания и значительное повышением КФК. Кроме того, в группе с дистальным типом Миоши у пациентов отмечаются миалгии, повышение АСТ и АЛТ, в связи, с чем эти пациенты длительное время наблюдаются и лечатся у терапевтов с диагнозами полимиозит и гепатит неясной этиологии.
При проведении молекулярно-генетического исследования в семьях с ПМДТМ и КППМД 2В выявлена только одна мутация в гене DYSF с. 573-574 ТG >АТ в гомозиготном состоянии с заменой р.Val67 Asp (валина на аспартат) в полипептидной цепочке [4,5].
Как известно, мутация: с.200_201 del ins AT ведет к замене в 67 позиции протеина аминокислоты валин на аминокислоту аспарагин (val167asp) в гене DYSF с заменой р.Val67 Asp.
Интерес представляет родословная семьи Р., где у пробанда (III,2) отмечаются клинические признаки ПМДТМ, возраст дебюта заболевания 19 лет. В неврологическом статусе: отмечается гипотрофические изменения в дистальных отделах предплечий и голеней, преимущественно задней группы, снижение сухожильных рефлексов с двуглавой и трехглавой мышц плеча, отсутствие карпорадиальных, коленных и ахилловых рефлексов с двух сторон. На момент осмотра КФК – 1373 Ед/л. ЭНМГ: первично-мышечный тип поражения. У троюродного сибса (III,6) клиническая картина соответствовала КППМД 2В, с дебютом заболевания в 21 год. На рис. 1 приведена родословная этой семьи.
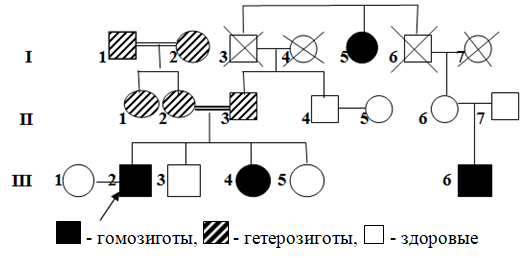
Рис. 1. Родословная семьи Р. ПМДТМ, АР тип наследования.
Нами было проведено молекулярно-генетическое исследование данной семьи, в ходе которого у больных членов семьи была выявлена мутация в гене DYSF. При этом у сибса (III,4) выявлена мутация в гомозиготном состоянии на доклинической стадии заболевания. Гетерозиготными носителями данной мутации явились родители пробанда (II: 2 и 3), бабушка и дедушка пробанда по материнской линии (I: 1 и 2), а также тетя пробанда по материнской линии (II: 1).
С учетом высокой распространенности КППМД в исследуемом горном изляте, было проведено молекулярно-генетическое исследование 80 фенотипически здоровых жителей горного изолята на наличие известной мутации (160 проанализированных хромосом). В результате проведенного исследования получены следующие результаты: у двух жителей выявлена мутация в гене DYSF, находившаяся в гомозиготном состоянии на доклинической стадии, еще у 23 фенотипически здоровых мутация находилась в гетерозиготном состоянии. Таким образом, частота гетерозиготного носительства мутации составила 14,38% (23/160).
Выводы:
- Проведенное в Республике Дагестан клинико-генетическое исследование ПМД выявило горный изолят с высоким риском развития КППМД 2В и ПМД дистального типа Миоши, что позволяет проводить диспансеризацию таких пациентов и эффективное медико-генетическое консультирование всем жителям данного изолята.
- Среди жителей горного изолята выявлены 23 фенотипически здоровых гетерозиготных носителя мутантного гена DYSF, что указывает на возможность проспективного медико-генетического консультирования в этих семьях и возможности проведения пренатальной диагностики.
- Среди жителей горного изолята выявлены пациенты с мутацией, находящейся в гомозиготном состоянии на доклинической стадии, что позволяет разработать медикосоциальные мероприятия в отношении прогноза их дальнейшего здоровья, включая проведение пренатальной диагностики, обеспечение психологической поддержки и социальной адаптации пациентов и членов их семей.
* Данный ген локализован на хромосоме 2 в области 2р13.3, состоит из 233141 нуклеотида, из которых 6246 нуклеотидов составляют 58 экзонов гена. В продуцируемом белке 2081 аминокислота