
Избыточный вес и ожирение, являясь компонентами метаболического синдрома (МС), прогрессивно нарастают в современном обществе, проявляя несомненную эпидемичность [5,20]. Известно, что к компонентам МС относят нарушение углеводного обмена (нарушенная гликемия натощак (НГН), нарушение толерантности к глюкозе (НТГ)), липидного обмена (комбинированная и изолированная дислипидемии – гипертриглицеридемия, гиперлипидемия липопротеинов низкой плотности (ЛПНП)), прогрессия аполипопротеинов (апоЛП) В и регрессия апоЛП-А, избыточный синтез сверхчувствительного С-реактивного белка, гиперинсулинемия (ГИ) и инсулинорезистентность (ИР) [11,16]. Исследования последних лет, посвященные изучению свойств класса адипокинов, позволили существенно расширить представления о патогенезе МС в целом и о явлении ИР в частности [3,5,15].
Однако вначале обратимся к рекомендованным критериям диагностики МС (ВНОК, 2009) [3]:
- основной признак – центральный (абдоминальный) тип ожирения – окружность талии (ОТ) более 80 см у женщин и более 94 см у мужчин,
- дополнительные критерии: артериальная гипертензия (АГ) (артериальное давление (АД)≥130/85 мм рт. ст.), повышение уровня триглицеридов (ТГ) (≥1,7 мМ/л), снижение уровня холестерина липопротеидов высокой плотности (ЛПВП) (<1,0 мМ/л у мужчин; <1,2 мМ/л у женщин), повышение уровня холестерина ЛПНП (>3,0 мМ/л), НГН (глюкоза в плазме крови натощак 6,1 мМ/л и более), НТГ (глюкоза в плазме крови через 2 ч после нагрузки глюкозой в пределах 7,8 и более и 11,1 мМ/л и менее) [2].
Наличие у пациента центрального ожирения и 2-х дополнительных критериев является основанием для диагностирования МС [2,7,20]. Жировой ткани принадлежит существенное значение в формировании МС и ассоциированными сосудистыми осложнениями [1,11,16]. Цитокины, продуцируемые жировой тканью (ЖТ), обладают способностью модифицировать инсулинопродукцию, изменяют трансдукцию инсулинового сигнала в периферических клетках, активность рецепторов к инсулину, интенсивность липогенеза [3,5,8,21]. Адипокины обладают бивалентной способностью в воздействии на ГИ и ИР. Так, накопление в крови фактора некроза опухоли альфа (ФНО-α) и его растворимого рецептора 1 типа проявляют ингибиторную активность в отношении синдрома ГИ и потенцирующую – при ИР [12,13,18].
Существенное значение в модификации ИР играют адипонектин (АН) и лептин, дополнительно оказывая влияние на изменение пищевого поведения [18, 19]. Продукт гена ADIPOQ (адипонектин, 30 kDa) – гормон, который синтезируется и секретируется белой ЖТ (преимущественно адипоцитами висцеральной области) и стимулируется инсулином. Показано, что АН тормозит дифференцировку преадипоцитов. Уровень АН в плазме крови обратно пропорционален массе ЖТ и индексу талия-бедро (ИТБ) [6]. АН регулирует энергетический гомеостаз и оказывает противовоспалительный и антиатерогенный эффекты [3,6,22]. Уровень АН снижается при ожирении в отличие от других повышающихся адипокинов, включая лептин, резистин и ФНО-α. Показано, что снижение экспрессии АН коррелирует с ИР, и АН влияет на метаболизм глюкозы [3,21]. АН выполняет протективную антигипергликемическую функцию, препятствует ИР и атеросклерозу, частично ингибируя активность ФНО-α[1,6,22]. ФНО-α – провоспалительный адипокин, повышенный уровень которого в плазме при ожирении связан с развитием гипергликемии и ИР, его способность подавлять экспрессию гена АН, возможно, основная причина снижения АН при ожирении [21,22]. У АН обнаружено ангиопротективное действие: снижение экспрессии молекул межклеточной адгезии, подавление пролиферации миоцитов сосудистой стенки и трансформации моноцитов в «пенистые» клетки, проявление эффекта снижения тромбогенного потенциала крови, интенсификация продукции оксида азота [1,8,13,21]. АН снижает системную воспалительную реакцию и способствует активации репарации клеток [12]. Было показано, что АН стимулирует активность керамидов, ассоциированных с рецепторами АН 1 и 2 типа [9,10], а также ускоряет их катаболизм и формирование антиапоптозных субстанций – сфингозин-1-фосфат – независимо от циклического аденозинмонофосфат-(цАМФ)-зависимой киназы (цАМФК). К первоочередным клеткам мишеням АН относят гепатоциты, кардиомиоциты, β-клетки поджелудочной железы, подоциты. Гиперпродукция АН жировой тканью ведет к улучшению периферической инсулиночувствительности, в то же время снижение активности и количества рецепторов АН повышает ИР [8,17,19]. Полагают, что активация цАМФК снижает формирование и активность рецепторов АН, однако механизм, определяющий подобную активность и стимулирование независимой цАМФК при АН-опосредованном улучшении гомеостаза глюкозы остаётся неясным. Керамиды индуцируют ИР через активацию протеинфосфатазы 2А и протеинкиназы С, усиливают системную воспалительную реакцию [17].
Лептин (белок, 16 kDa), кодируемый в жировых клетках геном, обусловливающим тучность. Концентрация лептина увеличена у пациентов, страдающих избыточным весом и ожирением [3,12,14,15]. Так показано, что снижение массы тела на 10% приводит к 53% снижению концентрации лептина. Напротив, 10% набор веса на 300% увеличивает уровень лептина, что может объясняться «лептинорезистентностью» – неспособностью лептина проникать к участкам связывания в гипоталамусе, отвечающим за регуляцию аппетита [14,15,22]. Показано, что лептин ингибирует секрецию инсулина, вызывая ассоциированное повышение гликемии [14]. Уровень лептина зависит от эндокринного гендерного статуса: концентрация лептина выше у женщин в сравнении с мужчинами. Высокий уровень лептина способствует прогрессированию ангиопатий и сердечно-сосудистых заболеваний (ССЗ), обусловленному тромбогенной активностью в результате особого взаимодействия между лептином и тромбоцитарными рецепторами к нему [22]. Показано, что изменение концентрации лептина коррелирует с динамикой проинсулина при синдроме ИР, проявляя прямую взаимосвязь. Это позволяет говорить о явлении лептинорезистентности как компоненте МС.
Другим более известным цитокином является ФНО-α и его комплексирующие растворимые рецепторы. ФНО-α рассматривается как провоспалительный цитокин, имеющий существенное значение в формировании системного воспалительного ответа при ряде соматических заболеваний [8,13,19]. Так показано, что ФНО-α избыточно синтезируется адипоцитами особенно у тучных пациентов, способствует гиперплазии клеток ЖТ, ингибирует цитокин-индуцированное поглощение и метаболизм глюкозы адипоцитами. Доказано, что гиперпродукция ФНО-α играет ключевую роль в патогенезе синдрома ИР, воздействуя на пути передачи инсулинового сигнала и активность рецепторов к инсулину [3,12,18,21]. Как ФНО-α, так и интерлейкин (ИЛ) 6 прямо коррелируют со степенью выраженности висцеральной ЖТ, степенью ИР и риском ССЗ. Гиперпродукция лептина и ряда других адипокинов у тучных пациентов индуцирует синтез мРНК ФНО-α и потенцируют его высвобождение макрофагальной системой [12]. Показано, что высокий уровень ФНО-α, лептина, ГИ в сочетании с ИР и недостатком АН существенно повышают риск развития осложнений при нарушении углеводного обмена и ССЗ. Контринсулярное действие ФНО-α опосредовано снижением экспрессии глюкозотранспортера (ГЛЮТ) 4 типа и ингибированием тирозинкиназы инсулиновых рецепторов в клетках-мишенях. Сочетанное влияние повышенного количества висфатина, ФНО-α, Ил-6 и недостатка АН, Ил-10, Ил-4 способствуют прогрессированию эндотелиопатии и ассоциированных осложнений [8,14,18].
Кроме того, показано, что уровень адипокинов сопряжен со степенью активности оксидативного стресса: снижение активности антиоксидантной системы ассоциировано с приростом ФНО-α, ИЛ-6, лептина и дефицитом АН [3,4]. Как оказалось, нарушение адипокинового баланса с превалированием ФНО-α, лептина и дефицитом АН на фоне ГИ и ИР модифицирует липидный обмен с увеличением холестерина ЛПНП и ТГ, компонентов «диабетической триады». Подобная форма дислипидемии существенно ускоряет атеросклеротический процесс и увеличивает риск ССЗ [7,11,12,21].
Материалы и методы исследования. Под наблюдением находилось 50 человек. Из них: женщин – 26 (52%), мужчин – 24 (48%). Средний возраст пациентов составил 47,9±0,64 лет, средняя длительность МС – 5,9±0,23 лет, средняя масса тела – 88,8±1,14 кг, значение индекс массы тела (ИМТ) – 30,7±0,37 кг/м2, ОТ – 98,1±0,97 см. Всем больным проводилась комбинированная терапия, включавшая гипокалорийное питание с учётом физической активности (1150-1350 ккал/сут), применение нутритивных корректоров, содержащих волокна клетчатки (мукофальк 10 г/сутки), инсулиносенситайзеров группы бигуанидов (средняя доза метформина пролонгированного действия – 1250 мг/сутки), индивидуальная физическая активность (плавание, ходьба с разной скоростью темпа, в режиме нарастающего тренда нагрузки). Лабораторные методы исследования рутинных параметров крови проводились по общепринятым методикам: препрандиальная гликемия натощак, уровень гликированного гемоглобина (HbA1c), прямым методом), общего холестерина (ОХС), холестерина ЛПНП и ЛПВП, ТГ, печёночных трансаминаз (как параметры контроля безопасности). У больных изучали следующие параметры адипокинового статуса: уровень ФНО-α (реактивы ООО «Цитокин» (Россия), адипонектина, лептина (реактивы «R&D System, Inc.» (США)) методом ELISA на анализаторе иммуноферментных реакций АИФР-01 «Униплан» («Пикон», (Россия)), также определяли базальный уровень инсулина.
Для каждого пациента на основе значений инсулина и глюкозы сыворотки крови препрандиального периода рассчитывали показатель HOMA по формуле (1):
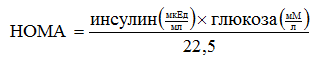 (1)
(1)
При базальном уровне инсулина ≥18 мкЕд/мл констатировалась ГИ, значения показателя HOMA≥2,27 свидетельствовал о наличии ИР [13,18,20]. Указанные параметры исследовались до начала терапии и спустя 3 мес. Статистическая обработка выполнена с помощью программ Excel 2010 (Microsoft) и Statistica 8.0 (StatSoft, Inc.), исследуемые показатели приведены в виде M±m, для сравнения использовали критерий Уилкоксона (W), критический уровень значимости (p) принимали равным 0,05.
Результаты исследования и их обсуждение. До начала терапии у всех пациентов отмечалась инсулино- и лептинорезистентность (уровень лептина составлял 31,0±1,04 нг/мл, инсулина – 18,4±0,40 мкЕд/мл, индекс НОМА – 5,19±0,12), а также сниженное значение АН – 3,97±0,06 нг/мл. Нарушение гликемического статуса соответствовало НГН, среднее значение гликемии натощак – 6,3±0,04 мМ/л, HbA1c – 6,17±0,07%. Значение уровня HbA1c обнаруживало прямую корреляцию с величиной инсулина (r +0,49, p<0,01), индекса НОМА (r +0,48, p<0,01), лептина (r +0,61, p<0,001), ФНО-α (r +0,59, р<0,001). Выявлена прямая зависимость инсулина и лептина (r +0,52, p<0,01), ФНО-α (r +0,87, р<0,001) и отрицательная корреляция с АН (r 0,59, p<0,001). Индекс НОМА проявлял схожую с уровнем инсулина тенденцию корреляции. Величина лептина отрицательно коррелировала с АН (r -0,36, p<0,01) и положительно – с ФНО-α (r +0,51, p<0,001). Обнаруживалась отрицательная взаимосвязь уровня ФНО-α и АН (r -0,46, p<0,001).
Анализ коррелятивных данных параметров липидного обмена выявил: положительную связь уровня ТГ с индексом HOMA (r +0,33, p<0,05), ЛПНП проявлял положительную корреляцию с HbA1c (r +0,52, p<0,001), инсулином (r +0,34, p<0,05), индексом НОМА (r +0,45, p<0,005), лептином (r +0,36, p<0,05) и ФНО-α (r +0,38, p<0,005), также обнаруживалась отрицательная связь ЛПВП с HbA1c (r -0,68, p<0,001), лептином (r -0,33, p<0,05) и ФНО-α(r -0,29, p<0,05).
Спустя 3 месяца от начала комплексной терапии масса тела снизилась на 8,9% (Δ-7,9 кг) и составила 81,0±1,19 кг, ИМТ уменьшился на 9% (Δ-2,7 кг/м2), достигнув 28,0±0,38 кг/м2, также отмечалось снижение ОТ на 8,8% (Δ-8,7 см), составившего 89,4±1,05 см (W, p<0,01). Уровень систолического АД снижался с 147±0,7 до 136±0,6 мм рт.ст. и диастолического АД – с 95±0,8 до 80±0,7 мм рт.ст. (W, p<0,05).
Параметры углеводного обмена характеризовались сходной тенденцией: уровень препрандиальной гликемии уменьшился на 16,9% до 5,3±0,07 мМ/л (Δ-1,0 мМ/л), HbA1c – на 17% до 5,1±0,04% (Δ-1,1%) (W, p<0,01). Значение инсулинемии снижалось на 53,8% до 8,5±0,13 мкЕд/мл (Δ-9,9 мкЕд/мл), индекса НОМА – на 61,8% до 1,9±0,03 (Δ-3,3) (W, p<0,01).
У пациентов отмечалось достоверное изменение показателей липидного обмена: уровень ОХС снижался на 22,9% до 5,4 мМ/л (Δ-1,6 мМ/л), ТГ – на 33,3% до 1,8 мМ/л (Δ-0,9 мМ/л), ЛПНП – на 26% до 3,7 мМ/л (Δ-1,3 мМ/л) (W, p<0,01).
Адипокиновый статус характеризовался следующими изменениями: величина лептина уменьшалась на 48,4% до 16±0,57 нг/мл (Δ-15 нг/мл), ФНО-α – на 42,5% до 6,9±0,09 пг/мл (Δ-5,1 пг/мл), в то время как концентрация АН демонстрировала рост на 181,6% до 11,2±0,20 нг/мл (Δ+7,2 нг/мл) (W, p<0,01).
Таблица 1. Модификация параметров клинико-лабораторного статуса у пациентов через 3 месяца от начала терапии

Примечание: * – уровень достоверности различий значений параметра р<0,05 при использовании критерия Уилкоксона
Комплексное воздействие модификацией суточной калорийности питания, индивидуализированной физической активности, нутритивной коррекцией мукофальком в сочетании с применением метформина оказывают существенное влияние на компоненты метаболического синдрома (коррекция массы тела, углеводного и липидного обмена) и способствуют как снижению инсулино- и лептинорезистентности, ингибированию синтеза ФНО-α, так и приросту адипонектина у наблюдаемых больных. Подобная модификация параметров внутренней среды, уменьшение массы тела и ИМТ способствуют снижению риска развития или прогрессирования ССЗ, формирования сахарного диабета 2 типа и ингибированию дальнейшей трансформации предиабетических состояний.