
Широкомасштабные исследования опухолевого генома показывают, что каждая опухоль специфична и поэтому может рассматриваться как индивидуальное заболевание, отличающееся по этиологии, прогнозу и терапии. Гетерогенны не только опухоли различного происхождения, но и злокачественные образования одного органа или ткани. Кроме того, даже опухоль одного пациента, начало которой дала одна клетка, к моменту обнаружения содержит различные популяции клеток, отличающихся по морфологии, экспрессии генов, метаболизму, подвижности, ангиогенному, пролиферативному, иммуногенному и метастатическому потенциалу [16].
Внутриопухолевая гетерогенность, по-видимому, является одним из значимых факторов, определяющих исход лечения. Неудовлетворительные результаты многих существующих методов объясняются тем, что они основаны на допущении, что все клетки и области опухоли одинаковы. Для повышения эффективности терапии необходимо учитывать изначальную гетерогенность опухоли и изменения в клеточном фенотипе, происходящие в процессе лечения, в результате адаптации опухолевых клеток к воздействию и селекции устойчивых клонов.
Причины и механизмы возникновения внутриопухолевй фенотипической гетерогенности пока мало изучены. Наиболее обсуждаемыми являются две модели: стохастическая, основанная на клональной эволюции опухоли, и иерархическая, базу которой составляют опухолевые стволовые клетки [8]. Эти две теории не являются взаимоисключающими, и, вероятно, оба механизма реализуются в процессе неопластической трансформация и прогрессии опухоли. Так или иначе, фенотип опухолевой клетки есть результат генетических и эпигенетических изменений, влияния микроокружения и приобретенной фенотипической пластичности [15].
Согласно современным представлениям, эволюция опухоли происходит скорее по ветвящемуся пути, чем по линейному. Многочисленные эксперименты доказывают сосуществование в первичной опухоли генетически различающихся клональных субпопуляций. Иногда выявляется значительная генетическая дивергенция между субклонами, что указывает на то, что они могут сосуществовать долгое время [15]. Исследования, направленные на реконструкцию пути, по которому эволюционирует опухоль, имеют важное значение. Они позволяют выделить гены, значимые для инициации и дальнейшей прогрессии злокачественного новообразования. Среди этих так называемых driver-генов могут быть новые биомаркеры и терапевтические мишени для целей диагностики и лечения онкологических заболеваний. Появление новых мощных технологий инициировали работы по секвенированию геномов опухолевых клеток. Целью этих исследований является выявление всех мутаций, характерных для каждого типа опухоли. Основная сложность такого анализа – отличить значимые для инициации и прогрессии опухоли мутации (driver-мутации) от случайных, накопленных в опухолевой клетке, но не критичных для канцерогенеза генетических изменений (passenger- мутации). Стандартным подходом в решении подобного рода проблем является поиск рекуррентных мутаций (или рекуррентно мутированных генов) в большой группе пациентов [19]
Светлоклеточная карцинома почки (СКП) – наиболее распространенная форма рака почки. Инактивирующая мутация в гене VHL, выявляемая почти в 80% случаев, является генетической особенностью этого типа рака. Мутация в этом гене довольно редко выявляется в опухолях других локализаций. При этом известные опухолевые гены, такие как RAS, BRAF, TP53, CDKN2A, PIK3CA, PTEN, EGFR и ERBB2, мутации в которых часто находят в различных карциномах, вносят незначительный вклад в развитие СКП [6]. Однако, сама по себе инактивация VHL не является достаточным условием для малигнизации [21]. Вероятно, должны произойти мутации и в других генах, чтобы клетка стала опухолевой. Недавно было показано, что у 41% пациентов с СКП выявляется инактивирующая мутация в гене PBRM1, кодирующем белок BAF180, который входит в состав комплекса, ремодулирующего хроматин [7]. Этот ген, также как и VHL, картирован в локусе 3р. Ранее был выявлен еще один ген, расположенный в этом же районе, мутации в котором обнаружены у 3% пациентов с СКП. Это ген SETD2 (метилтрансфераза H3K36). По-видимому, модификация хроматина и гистонов играет значимую роль в патогенезе СКП, о чем свидетельствуют выявляемые в опухолях соматические мутации и в других генах (KDM6A, KDM5C), которые также как PBRM1 и SETD2 вовлечены в регуляцию структуры хроматина [13].
Результаты работы Swanton c соавторами показали, что опухоли СКП генетически гетерогенны [9]. Используя различные молекулярно-генетические методы, у четырех пациентов были проанализированы образцы ДНК и мРНК, выделенные из разных частей опухоли, а также метастазов. Высокий уровень гетерогенности был выявлен даже среди соседних образцов. Было показано, что 63-69% из обнаруженных мутаций являются уникальными и не присутствовуют в других областях опухоли. Из всех генов, ассоциированных с СКП, только VHL имел одинаковые мутации во всех образцах. При этом у разных пациентов они были разными. Неожиданным оказался характер мутаций в генах SETD2 и KDM5C. В разных частях опухоли одни и те же гены имели разные мутации, но все эти мутации приводили к потере функции. Это свидетельствует о конвергентном характере эволюции опухоли почки. Транскрипционный анализ показал, что в одной той же опухоли присутствуют субклоны, которые по экспрессионному профилю относятся к разным прогностическим группам. Из этого следует, что исследование только одного биопсийного образца, может оказаться не информативным и не отражать состояние опухоли. Высокая фенотипическая гетерогенность значительно усложняет и задачу выбора тактики лечения. Реконструкция опухолевой клональной архитектуры и поиск ключевых мутаций, которые возникли на ранних стадиях развития опухоли и присутствуют таким образом во всех опухолевых клетках и метастазах, может оказаться полезным в целях поиска надежных биомаркеров и эффективных путей терапии.
Таким образом, кандидат на роль driver-гена СКП должен удовлетворять следующим требованиям: 1 – изменение экспрессии такого гена должно часто встречаться среди пациентов с СКП; 2 - изменение активности гена должно происходить уже у пациентов c начальной стадией заболевания; 3 – изменение экспрессии гена должно выявляться во всех частях опухоли.
Одним из таких генов может быть DUSP9/MKP4, протеинфосфатаза МАР киназ (МКР) из семейства биспецифических протеинфосфатаз. МКР инактивируют МАР киназы, дефосфорилируя тирозиновые и серин/треониновые остатки аминокислот в активирующем сайте [4]. Подавление транскрипции DUSP9 в опухолевой ткани у большинства пациентов с СКП было показано в наших работах [1,2] и в работах других исследователей [3,20]. Значительное снижение активности гена выявляется уже на начальных стадиях заболевания. Широкомасштабный анализ, проводимый в рамках международного проекта Сancer Genome Project, направленного на поиск мутаций в экзонах генов в различных опухолях, не выявил в СКП генетических изменений в кодирующей последовательности DUSP9 [6]. Механизмы транскрипционного подавления пока не изучены. В работе [2] нами было показано, что в регуляцию экспрессии могут быть вовлечены эпигенетические механизмы, в частности метилирование промоторной области. Не исключено, что причиной инактивации могут быть соматические мутации в регуляторной области. Мы секвенировали промоторные участки в двух образцах, в которых ранее не выявили метилирования. Отсутствие мутаций предполагает, что возможны и другие пути инактивации, например, путем модификации структуры хроматина. Активное участие этого механизма в канцерогензе почки подтверждают экспериментальные данные [13].
В представленной работе мы сравнили уровень экспрессии гена DUSP9 в разных частях опухоли почки и в прилежащей морфологически неизмененной (условно нормальной) ткани пациентов с СКП, используя метод полуколичественной полимеразной цепной реакции, сопряженной с транскрипцией (ОТ-ПЦР).
Материалы и методы
Опухолевые образцы и выделение РНК. Первичные опухоли и прилежащие к ним условно нормальные ткани почек были предоставлены отделением Эндоваскулярной урологии ФБГУ РНЦ РХТ (г. Санкт-Петербург). После нефрэктомии материал был заморожен в жидком азоте и хранился до использования при - 800 С. Все образцы прошли патоморфологическое исследование и были охарактеризованы.
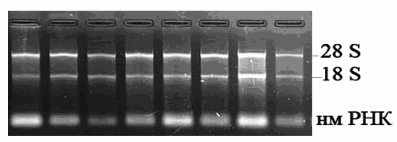
Из трех разных удаленных друг от друга частей опухоли и нормальной ткани отбирали образцы размером ~ 1 см3 , из которых выделяли тотальную РНК стандартным гуанидин-тиоцианатным методом. Для этого клетки лизировали в 4М гуанидинтиоцианате (pH4) и экстрагировали РНК кислым фенолом. Количество и чистоту препаратов РНК оценивали по соотношению А260/А280. Качество (интактность и присутствие ДНК) проверяли методом электрофореза РНК, денатурированной глиоксалем.
ОТ-ПЦР. кДНК синтезировали в 25 мкл реакционной смеси, содержащей 2 мкг тотальной РНК, 0.5 мкг олиго (dТ)12-18, 200 ед обратной транскриптазы MMLV («Promega»), по 500 мкМ каждого дНТФ, 25 ед ингибитора РНКаз («Promega»), 5 мкл 5х буфeра для обратной транскриптазы. Реакцию проводили 1,5 часа при 420С, затем прогревали смесь 3 мин при 800С, добавляли еще 200 ед. обратной транскриптазы и инкубировали реакционную смесь дополнительно 1 час при 420С.
Реакцию ПЦР проводили с использованием набора для ПЦР фирмы «Litex» в объеме 25 мкл. Реакционная смесь содержала 1мкл кДНК, 400 нМ каждого праймера, 200 мкМ каждого dNTP и 1.5 мМ МgCl2. Амплификация фрагмента гена DUSP9 проводилась в присутствии 5% (по объему) DMSO. Реакцию проводили в амплификаторе AB 9700 (Applied Biosystems) по следующей программе: для бета-актина: 950С , 1 мин; затем 25циклов- 940С, 30 сек; 640С, 1 мин; 720С, 2 мин. Заключительный цикл – 720С 8 мин. Для DUSP9: 950С , 3 мин; затем 35циклов - 940С, 1 мин; 600С, 50 сек; 720С, 2 мин. Заключительный цикл – 720С 8 мин.
Праймеры, использованные в работе:
бета-актин: 5’-TGGCACCACACCTTCTACAA-3’ (прямой) и
ACAGCGAGGCCAGGATGGAG-3’ (обратный);
DUSP9: 5`-CCT GTG GCT GCG TCG GGA GCT GTC-3` (прямой) и
5`-GAC CCC CGC CAA GCA GTG GAC GAG -3` (обратный);
Продукты ПЦР анализировали в 1.2%-ном агарозном геле.
Для количественной оценки уровня экспрессии генов в опухолевой и нормальной тканях использовали цифровую фотодокументационную систему и программное обеспечение EDAS 290 (Kodak Digital Science)
Результаты и обсуждение
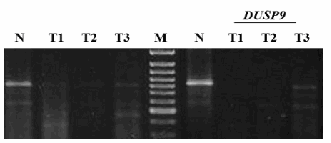
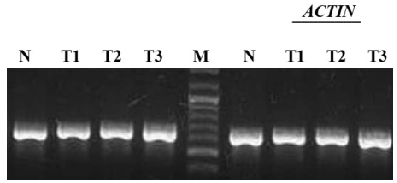
Опухоли светлоклеточной карциномы почки имеют высокий уровень фенотипической гетерогенности, что является следствием накопления мутаций (как генетических, так и эпигенетических) в процессе эволюции опухоли. Мы исходили из того, что мутации, которые произошли на ранней стадии и дали клетке селективное преимущество, будут сохранены во всех последующих субпопуляциях, и поэтому должны выявляться во всех клетках опухоли. Для этого мы оценивали уровень экспрессии гена DUSP9 в трех разных участках опухоли у четырех пациентов со стадиями I и II (по классификации TNM). У всех пациентов во всех участках опухоли уровень мРНК DUSP9 значительно ниже, чем в нормальной ткани (рис.1).
 |
 |
| Рис. 1 (а) | Рис. 1 (б) |
 |
|
| Рис. 1 (в) |
Рис. 1. а - РНК, выделенная из образцов; нм РНК – низкомолекулярная РНК; б, в - Результаты ОТ- ПЦР с указанными праймерами; N – нормальная ткань; Т – опухолевая ткань.
Мы пока не знаем причины инактивации, но важно то, что, по-видимому, такой фенотип обеспечивает клеткам селективное преимущество и возникает рано, поскольку уже в опухолях пациентов с начальной стадией наблюдается подавление транскрипции. Интересно отметить, что поскольку DUSP9 локализован на Х хромосоме (Хq28), то для его инактивации достаточно одного события ( на единственной Х хромосоме у мужчин и активной Х хромосоме у женщин).
Как правило, driver-мутации происходят в генах, вовлеченных в клеточные сигнальные и регуляторные пути [19]. DUSP9 относится к группе цитоплазматических МКР с широким спектром субстратной специфичности (Erk> p38 >JNK) и узким спектром экспрессии: DUSP9 экспрессируется преимущественно в плаценте, эмбриональной печени, а постнатально главным образом в почке [5, 17]. Какова биологическая роль DUSP9 в почке пока не известно. Данные, полученные за последние годы, показали, что МКР являются ключевыми молекулами, которые контролируют и регулируют активность MAP-киназных каскадов [10, 12]. Эти сигнальные пути вовлечены во все основные процессы, включая дифференцировку, пролиферацию, апоптоз [18]. Поэтому, можно полагать, что инактивация DUSP9 может приводить к нарушению основных процессов, обеспечивающих нормальную работу клеток эпителия почки.
Экспериментальные доказательства ассоциации ранних этапов неопластической трансформации с подавлением экспрессии DUSP9 были получены в работе [14]. Авторы изучали процесс канцерогенеза клеток мышиного эпидермиса и показали, что инактивация DUSP9 является инициирующим событием на пути прогрессии эпидермальных клеток к более инвазивному и метастазирующему фенотипу. Восстановление экспрессии DUSP9 в опухолевых вызывала их гибель. Особо следует отметить следующий факт: в данной модельной системе канцерогенеза опухолевая трансформация эпидермальных клеток происходит без активирующей мутации в генах семейства RAS. Соматические мутации в RAS-генах, и как следствие, повышение активности МАР киназ, довольно часто выявляются при онкопатологиях различного происхождения, однако практически не встречаются при раке почки. При этом аберрантная активация MAPK-сигнального пути выявлена на разных стадиях канцерогенеза почки, включая малигнизацию, ангиогенез и метастазирование [11]. RAS-ERK сигнальный путь регулирует процессы пролиферации и выживания клеток. Возможно, в опухоли почки инактивация DUSP9, негативного регулятора ERK, может приводить к разбалансировке этого сигнального каскада.
Таким образом, опухолевосупрессорные функции DUSP9 и ранняя его инактивация в процессе канцерогенеза свидетельствует о том, что он может быть driver-геном, инициирующим развитие СКП.
Работа выполнена при финансовой поддержке Минобрнауки России в рамках ФЦП «Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России на 2007-2013 годы» с использованием оборудования ЦКП «Биотехнологический центр исследования экспрессии генома» ФГБУ РНЦРХТ МЗСР России в рамках государственного контракта № 16.552.11.7021.